README
Please Read this document on gitbook: https://wangbch.gitbook.io/soapml-document/
soapml
This is a Chemist-Friendly tool, just very simple python code to test if this tool is useful to your work, no extra machine learning knowledge is needed.
soapml is based on SOAPLite: https://github.com/SINGROUP/SOAPLite
A machine learning tool for doing regression using SOAP (smooth overlap of atomic position) encoded structure of molecules, surface, ... Helps to find relationship between position and energy, activity and other physical chemical property.
How to install?
Test environment is Python3. Anaconda3 is highly recommended
Install SOAPLite: https://github.com/SINGROUP/SOAPLite , if you can not install it on Windows, use linux.
Install my machine learning tools: https://github.com/B-C-WANG/MachineLearningTools (download files and run "python setup.py install")
Install my tools for extracting information from Vasp dirs: https://github.com/B-C-WANG/VDE-VaspDataExtract (download files and run "python setup.py install")
Install soapml: download files and run "python setup.py install"
Demo
Train & Test
# prepare data
data = ...
# a vasp_file_path, list of string, like ["\public\Pt_OH1","\public\Pt_OH2"]
vasp_file_path, y = data
# make dataset
dataset = Dataset.from_vasp_dir_and_energy_list(vasp_file_path,final_ads_energy=y,
description="""
the carbon nanotube data,
doped with N or B,
adsorbate: OH
""")
# delete first 15% sample in each vasp dir (a vasp dir is a sample group)
dataset.sample_filter(ratio=0.15)
# apply period on VectorC, it is Z direction in this case, repeat +Z, 0 and -Z
dataset.apply_period(direction=2,repeat_count=1)
dataset.soap_encode(center_atom_cases=[8],encode_atom_cases=[5,6,7])
# save for loading later
dataset.save("dataset.smld")
# do machine learning
dataset = Dataset.load("dataset.smld")
model = Model(dataset)
# use gradient boost regression
model.fit_gbr(n_estimators=200,shuffle=True,test_split_ratio=0.3)
model.save("model.smlm")Result: average error 0.027, fig of model predicted_y and true_y

Validate
# load trained model
model = Model.load("soapmlGbrModel_test.smlm")
# offset a new sample_group, from vasp dir
validate_vasp_dir_path = ...
final_ads_energy = ...
dataset = Dataset.from_vasp_dir_and_energy_list(
vasp_dirs=[validate_vasp_dir_path],
final_ads_energy=[final_ads_energy],)
# use model.encode, will encode the same way as dataset \
# when create Model (the dataset of Model(dataset))
dataset = model.encode(dataset,center_atom_cases=[8],sample_filter_ratio=0.15)
model.predict_and_validate(dataset)Use
# load model
model = Model.load("soapmlGbrModel_test.smlm")
# use a existing vasp dir, but not offer y
vasp_dir_index = 5
dataset = Dataset.from_vasp_dir_and_energy_list(
vasp_dirs=[x_file_path[vasp_dir_index]],
only_x=True)
# obtain a slab_structure from exsiting vasp samples
slab_structure = dataset.give_a_sample_from_dataset(sample_group_index=0,
sample_index=-1,
use_repeated=False)
# use the same box_tensor
box_tensor = dataset.box_tensor
# use custom center_position
center_position = np.array([[0,0,0],[-10,-10,-10],[5,5,5]])
# make a new dataset for predict
dataset =Dataset.from_slab_and_center_position(slab_structure=slab_structure,
center_position=center_position,
box_tensor=box_tensor)
# encode the same way as dataset in model
dataset = model.encode(dataset, center_position=center_position)
print(dataset.datasetx)
# predict y and output
pred_y = model.predict(dataset)
print(pred_y)Other Demo - Coming Soon ...
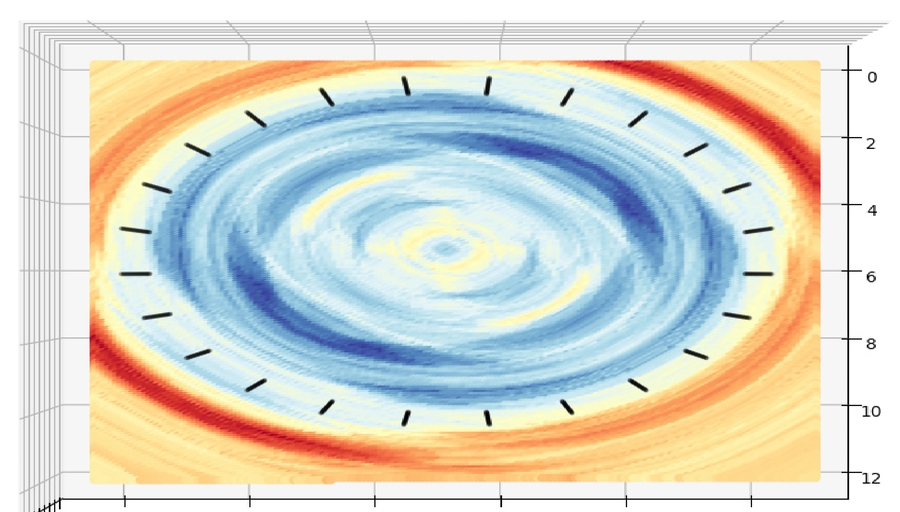
And we can predict the energy in any position, here we predicted OH* adsorption energy on a plane, where z is equal to the z-coordinate of O atom:

What can soapml do?
By training the data that contains the relation ship of e.g., position-energy, soapml can predict the energy on any position, make it possible to find some position with higher or lower energy.
Can I use soapml now?
soapml is now supported for ΔE prediction of a background structure (slab) with an active small group (ads). It should contain only one ads on the slab, and the position of ads has a big influence on ΔE, and ΔE should be E(slab+ads) - E(slab).
How to use soapml? -- Make input as dataset
You can make your input by soapml.Dataset, soapml needs structure (atom index and x y z coordinate) and energy as input. It is now supported for VASP results dir (the dir containing OUTCAR, CONTCAR, OUT.ANI of VASP), and directly use structure array list and energy array list. You can write something in param "description" to help you remember what the dataset is. You can use one of following method to make your input, they all return an instance of Dataset, and if you want to make only dataset of X (when do prediction, you need to make dataset contains only X), you need to give only_x = True.
——————————
1. @static - Dataset.from_vasp_dir_and_energy_table
(vasp_dir_table, only_x, description)
First, give a dir containing many vasp dirs, these dirs have slab + ads structure. Use @ static - Dataset.generate_vasp_dir_energy_table(vasp_dir, to_csv), to generate a excel or csv table, you need to fill the table with slab energy of every vasp dir, like:
Vasp Dirs
slab energy
Pt_OH1
1.0
Pt_OH2
2.0
Pt_OH3
3.5
Then use @static - Dataset.from_vasp_dir_and_energy_table(vasp_dir_table,description) and set vasp_dir_table to the excel or csv filename.
——————————
2. @static - Dataset.from_vasp_dir_and_energy_list
(vasp_dirs, slab_energy, final_ads_energy, only_x, description)
vasp_dirs is a list of string containing your vasp dir path, slab energy or final_ads_energy is list that have same length of vasp_dirs. You can only give either slab_energy or final_ads_energy:
Regression target Et will be: energy of every step - slab_energy
Regression target Et will be: energy of every step - energy of final step + final_ads_energy.
——————————
3. @static - Dataset.from_coordinate_and_energy_array
(coordinate, energy, box_tensor, only_x, description)
Considering different samples may contain different atom number, the coordinate and energy are List of Array. The list length is n_sample_group, for each sample group, coordinate is an Array with shape (n_sample, n_atom, 4), and energy is an Array with shape (n_sample), the 4 means [atom_index, x, y, z], atom_index can be obtained from the periodic table (H:1, C:6, O:8, ...).
Box_tensor, or translation vector, is related to period structure, it can be seen as [VectorA, VectorB, VectorC] of the cell. The box_tensor should be a list of 3x3 arrays, since different sample_group may contain different cell, you have to offer different 3x3 arrays for each sample_group.
Box_tensor can be directly extract from POSCAR in vasp or Material Studio.

When you finished your input, you will get an instance of Dataset as return, and now you need following functions to do SOAP transform to prepare x and y for machine learning:
——————————
4. @static - Dataset.from_slab_and_center_position
(slab_structure, center_position, box_tensor)
Only need to feed a (n_atom, 4) array as slab_structure, a (n_center_position, x) array center_position and a list of 3x3 (most possible an one-element list) array box_tensor.
——————————
5. Dataset.give_a_sample_from_dataset
(sample_group_index, sample_index, use_repeated)
Return a coordinate with shape (n_atoms, 4), if repeated is true, use the structure after apply_period(). The coordinate if from the sample in sample_group_index + 1 th element and the sample_index + 1 th frame.
——————————
6. Dataset.sample_filter
(ratio)
Take ratio = 0.15 for example, the first 15 % of samples in each sample group will be deleted. It helps we some sample are unstable in vasp results.
——————————
7. Dataset.apply_period
(direction,repeat_count)
For periodic structure, we need to repeat structure. Direction = 0, 1, or 2, means we will add VectorA, B or C (in box_tensor) into all coordinates, repeat_count means how long we need to repeat.
When do we need to apply period? If the position we interested is very closed to the edge of cell, we need to repeat, also, if we set a large cut off, there may not be enough atom, we need to repeat (e.g., our cell have a length of 0.5 nm, but cutoff is 1.2 nm, we need to repeat twice to make length to 1.5 nm.)

——————————
8. Dataset.soap_encode
(center_atom_cases, encode_atom_cases, n_max=8, l_max=8, r_cut=15.0, absent_atom_default_position, relative_absent_position)
In SOAP, we need to give a position that are highly related to energy.(e.g. the position of O* is important to OH* adsorption energy on Pt(111)) So how do we find the position? One way is to set the position by calculation the average position of atoms which have index in center_atom_cases.
ONLY position of atoms in original structure are allowed to be used to get center position (repeated atoms are only for local environment feature.)
n_max=8, l_max=8, r_cut=15.0 are parameter of SOAPLite
And for encoding local environment, some atoms should not be included, e.g. if we use position of OH* (on Pt(111)) as center position, its environment should not contain OH* itself, so encode_atom_cases should be [78] rather than [1,8,78]. But what if there's no atom in encode_atom_cases? These absent atoms' position will be set into absent_atom_default_position, but there are two ways to set that position, you can switch it by setting different bool value of relative_absent_position:
The position of atom not in encode_atom_cases will be set into absent_atom_default_position
The position of atom not in encode_atom_cases will be set into the first center_position - absent_atom_default_position
Usually we should set the position of absent atom as far away from center position as possible, and the default value is [-10,-10,-10] off the center position. But in my case, set [-10, -10, -10] got a much better result than set to [-30, -30, -30], don't know why this happen.
——————————
9. Dataset.save
(filename)
Dataset will be saved to soapmlDataset file (.smld) using pickle. It's highly recommended to save your dataset and load it before train a machine learning model, since it takes to much time to do SOAP encoding.
——————————
10. @static - Dataset.load
(filename)
Load dataset instance from the soapmlDataset file (.smld).
How to use soapml? -- Use dataset to do machine learning
use soapml.Model and give a soapml.Dataset to train model, test model and save model. There are 4 stages to use soapml.Model, and their process are:
Make a Dataset containing X and Y, and encode them
Offer the Dataset in Model, and fit
Make a Dataset containing X and Y, but do not encode them
Load a trained model from Model.Load
Use Model.encode_before_predict to give the dataset and encode with the same config as in train process
Run Model.predict and offer the encode Dataset, will return Y and plot out predicted Y - true Y
Make a Dataset containing only X, and this X must be a list of array (Vasp results dir input is not supported -- since you have done using vasp, you need not to do soapml)
Load a trained model from Model.Load
Use Model.encode_before_predict to give the dataset and encode with the same config as in train process
Run Model.predict and offer the encode Dataset, and this time you have other ways to make center position.
——————————
1. Model.init
(dataset)
You need to offer a soapml.Dataset to create model. You can feed a dataset with both X and Y or only X (remember to set only_x = True).
——————————
2. Model.keep_data_larger_than & Model.keep_data_smaller_than
(y)
Keep data that have y larger/smaller than a threshold.
——————————
3. Model.fit_gbr
(test_split_ratio, n_estimators, shuffle)
Use gradient boost regression (gbr) model to fit x and y in dataset, error and a fig of predicted_y - true_y will print out. test_split_ratio means we use how many dataset to train and test (e.g., 0.3 means we use 70% data to train and use 30% data to test.), n_estimators is the parameter of gbr, shuffle is a bool, which means if we shuffle the dataset or not.
——————————
4. Model.encode
(dataset, center_atom_cases, center_position, sample_filter_ratio)
Encode the input dataset the same way as the dataset in Model.init, and return encoded dataset. You can feed only one of center_atom_cases and center_position.
——————————
5. Model.predict_and_validate
(dataset)
Use the X in encoded dataset to predict Y, plot pred_y - true_y and give out error.
——————————
6. Model.predict
(dataset)
Use X encoded dataset to predict and return y.
——————————
7. Model.save
(filename)
Model will be saved to soapmlModel file (.smlm) using pickle. Save and keep your model well, one day it will be useful.
——————————
8. @static - Model.load
(filename)
Load model instance from the soapmlModel file (.smlm).
Advance Example - SOAP "Probe"
prepare a surface and an adsorbate (e.g. H) as "Probe".
get energy of surface without probe Es, move probe on the surface, get energy list Ep_l. (from DFT calculation...)
now you have structures of different probe position on surface as X, Ep_l - Es as y
use SOAPTransformer to transform X to features, the position of probe is as center position
use ML models, like Gradient Boost Regression. Use soap features and y to train the model, save the model
give a new surface, give many center positions, use SOAPTransformer to get features, and used saved gbr model to give energy.
plot out.
e.g. use atom H as probe, nanotube as surface, calculate absorbation energy of H, got dataset, and use model to predict energy on a plane on the center of nanotube(red is high energy, blue is low energy):
Last updated
Was this helpful?